DOCUMENTATION vSampler webserver help Input formats Options Basic options Advanced options Other options Outputs vSampler local version Quick Start System requirement Download Test vSampler Sampler Examples Outputs Detailed options Other vSampler utilities (for advanced usages) BuildDatabase BuildIndex vSampler Database Database construction Annotation database Sampling database Number of variants in vSampler database Annotation of variant properties FAQ Links Cite vSampler
vSampler webserver help
Input formats
Web-based vSampler supports five types of files as input, including rsID, VCF, VCF-like, Coord-Only, Coord-Allele.
Note
All format should be
Users can paste plain text and uploaded file(
The upload limit is
Options
Basic options
- Population (default: EUR) : Sampling variants based on one of the 1000 genome phase3 super populations {EUR, EAS, AFR, AMR, SAS} genotype data.
- Minor Allele Frequency Deviation (default: +/- 0.05) : Maximum allowable deviations for minor allele frequency.
Advanced options
- DTCT Deviation (default: null) : Maximum allowable deviations for distance to closest transcription start site.
- Gene Density Deviation (default: null) : Maximum allowable deviations for gene density. Users can choose LD thresholds using r² > {0.1, 0.2, ..., 0.9} or Physical Distance thresholds from 100kb to 1000kb.
- Number of Variants in LD Deviation (default: null) : Maximum allowable deviations for the number of buddy variants in LD at various r² thresholds in r² > {0.1, 0.2, ..., 0.9}.
- GC Content Deviation (default: null) : Maximum allowable deviations for the GC Content around the variant using window size {100bp, 200bp,...,500bp}.
- Match Epigenomic Marks (default: null) : Whether to sample control variants with the same annotation of cell type-specific epigenomic mark.
- Match eQTL Significance (default: null) : Whether to sample control variants with the same GTEx v8 eQTL significance (significant/not significant). Select 1 of 54 tissue.
- Match Coding/Noncoding Region (default: null) : Whether to sample control variants falling in the same region (coding/noncoding).
Other options
- Sample Across Chromosome (default: false) : Indicator of sampling across chromosomes or not. Note: this option might increase the runtime by 2-4 times.
- Exclude Input SNPs (default: true) : Indicator to exclude input variants from matched variants or not.
- Match Variant Type (default: true) : Whether to sample control variants with the same variant type (SNP/INDEL).
- Sampling Number (default: 1) : The number of controls for each query variant.
- Annotation Number (default: 1) : When `Sampling number` is large, the control variants and its annotation information will make the output file extemely large. Thus it's necessary to output the annotation information of only a subset of control variants. This number is defined by `Annotation number`.
- Random Seed (default: NA). : Set random seed to ensure reproducibility.
Outputs
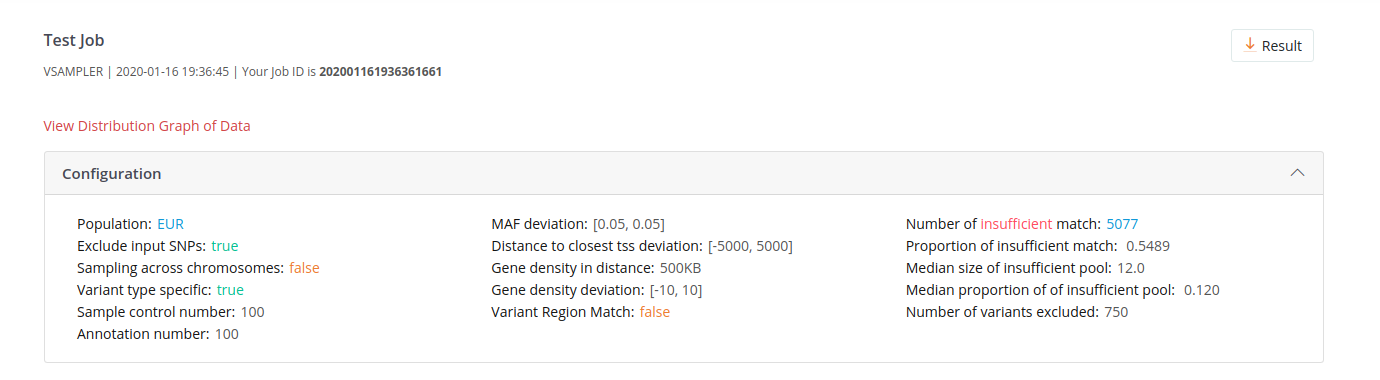
Configuration
shows all configuration of the submitted job
Prioritization Table

Results of vSampler will be displayed in tabular form, which can be easily sorted and filtered.
- Basic job information including job name, submission time and job id.
- Show unannotated query or Show insufficient match records to the table.
- Results, background of query records are colored in aquamarine, and the first column of this table is the label followed by sampling ratio in parentheses. Sufficient records are marked with green √
- Display configurations, users can define number of records to display, and filter the data use
Filter Setting
Note
Due to the web page capacity, this table will show up to 5 controls for each query, users can download the results to local machine and check all the results.
Distribution Graph of Data

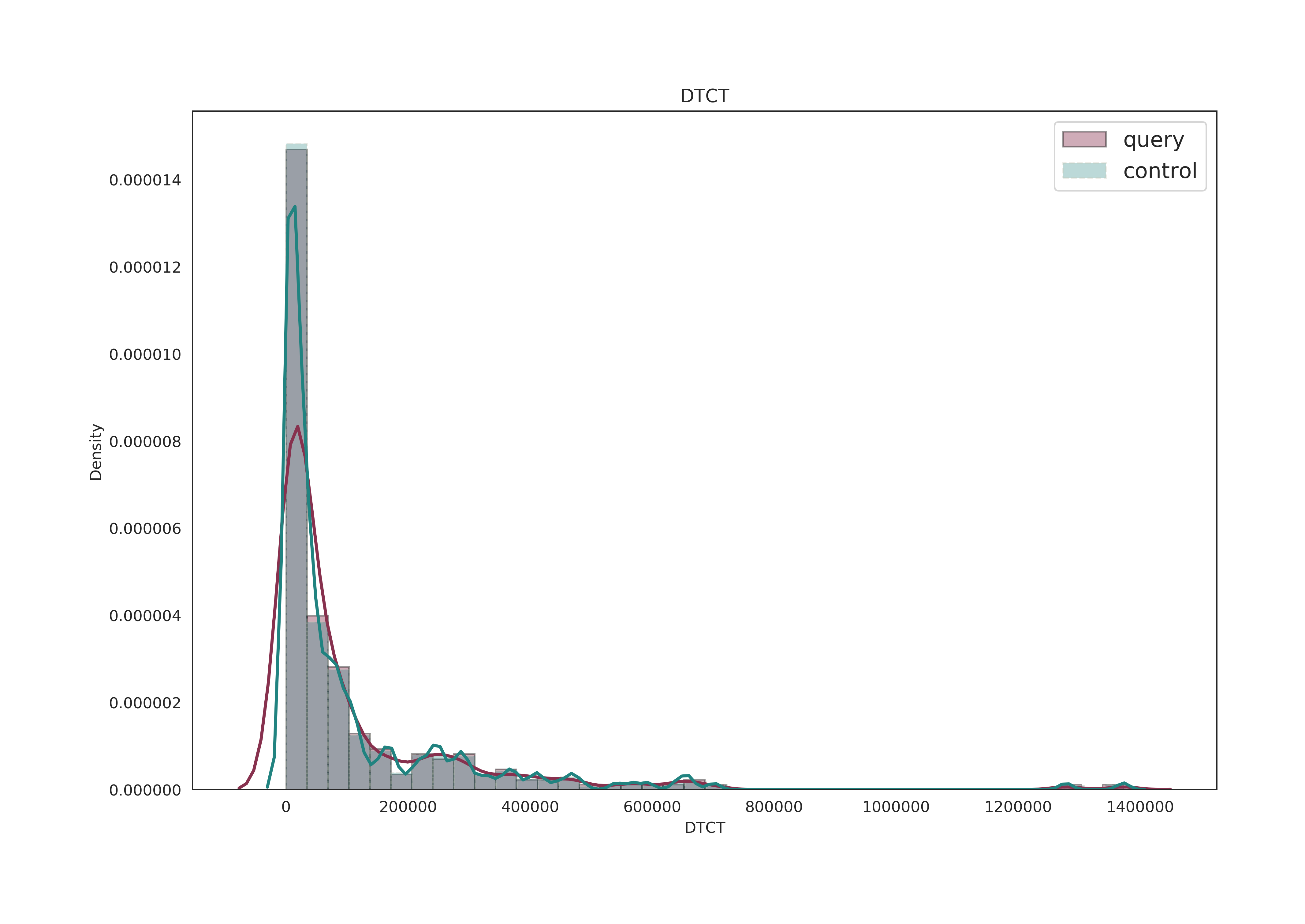
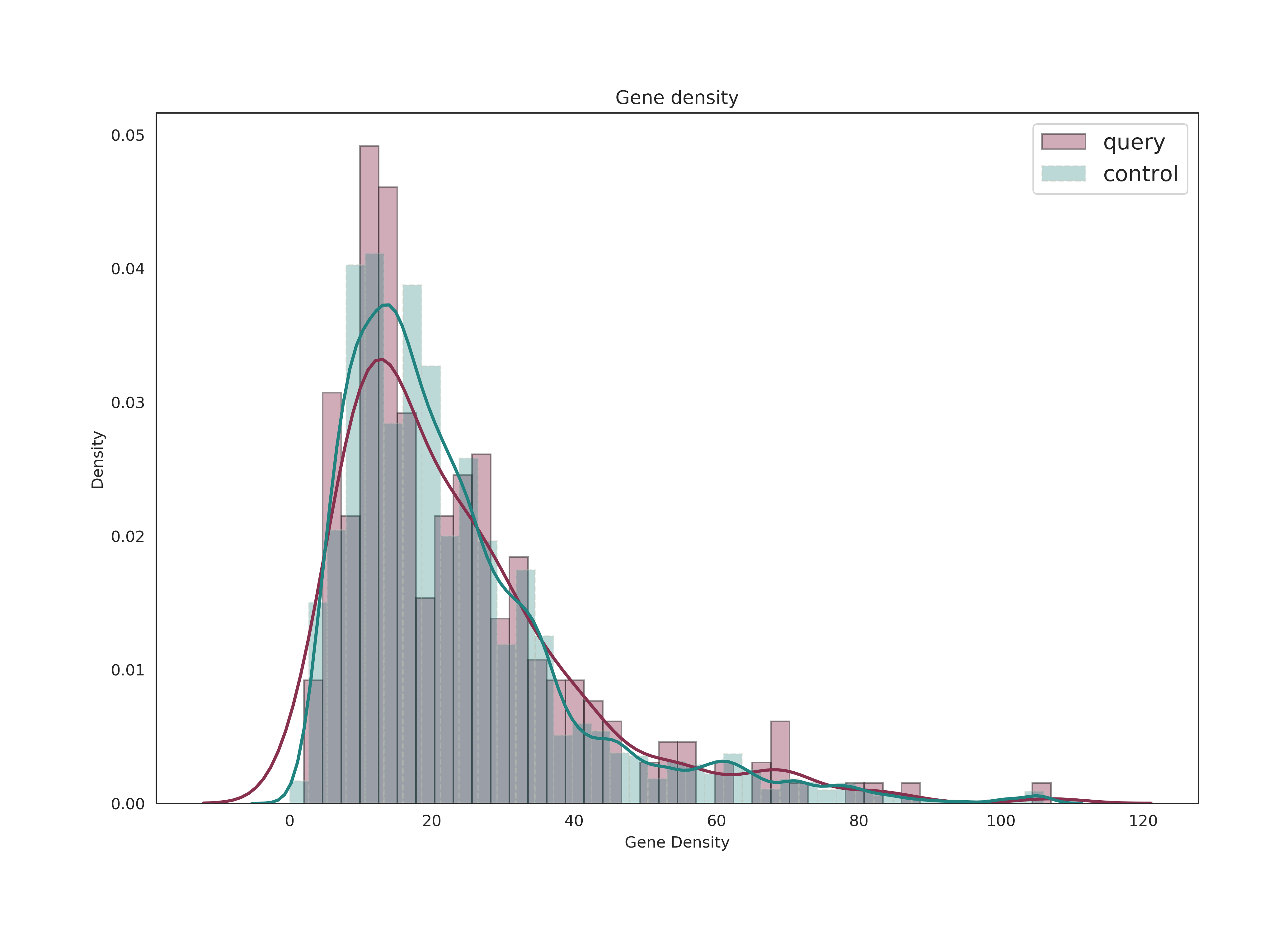


Reports the density distribution of MAF, DTCT, Gene Density, LD variants number, GC content.
vSampler local version
Quick Start
System Requirements
Java Runtime Environment (JRE) version 8.0 or above is required for vSampler. It can be downloaded from the Java web site. Installing the JRE is very easy in Windows OS and Mac OS X. In Linux, you have more work to do. Details of the installation can be found see the http://www.java.com/en/download/help/linux_install.xml.
Download
This steps guides you from downloading the vSampler program.
- The latest version of vSampler can be downloaded from https://github.com/mulinlab/vSampler/releases.
- Extract the ZIP archive, you should find file called VariantSampler-x.y.z.jar which can be used for run vSampler.
- You should also download indexed genotype reference panels from https://1drv.ms/f/s!Aurnn0fjCLv3glA_EoGK_N-daIwo?e=rKm9bh
vSampler sample variants based on 1000G phase 3 genotype data of 3 super populations including
Note
Our built up database using
User could build new database with other
Test vSampler
To test that you can run vSampler tools, run the following command in your terminal application, providing either the full path to the VariantSampler-x.y.z.jar file:
java -jar -Xms1g -Xmx4g VariantSampler-x.y.z.jar
You should see a complete list of all the tools in the vSampler toolkit.
USAGE: java -jar /path/to/VariantSampler-x.y.z.jar <program name> [-h]
Available Programs:
--------------------------------------------------------------------------------------
Database: Database build related.
BuildDatabase Build Database
BuildIndex Build Index
--------------------------------------------------------------------------------------
Sampler: Sampler related.
Sampler SamplerThe arguments
Sampler Examples
- Local vSampler requires query file and indexed genotype reference panels as inputs.
- Local vSampler supports five types of query file, including VCF, VCF-like, Coord-Only, Coord-Allele, TAB.
java -jar -Xmx4g -Xms1g VariantSampler-x.y.z.jar Sampler -Q:vcf data/example.vcf -D data/EUR.gz
java -jar -Xmx4g -Xms1g VariantSampler-x.y.z.jar Sampler -Q:vcfLike data/example.vcflike.tsv -D data/EUR.gz
java -jar -Xmx4g -Xms1g VariantSampler-x.y.z.jar Sampler -Q:coordOnly data/example.coordOnly.tsv -D data/EUR.gz
java -jar -Xmx4g -Xms1g VariantSampler-x.y.z.jar Sampler -Q:coordAllele data/example.coordAllele.tsv -D data/EUR.gz
java -jar -Xmx4g -Xms1g VariantSampler-x.y.z.jar Sampler -Q:tab,c=2,b=3,e=4,0=true data/example.tab.tsv -D data/EUR.gz
Outputs
vSampler will generate a zip containing the following four files:
- anno.out.txt - This file contains the annotations of query and controls. Example Data.
- sampler.config.txt - This file contains the configuration of the job, (i.e. the parameters used when running vSampler). Example Data
- sampler.out.txt - This file reports the sampling outputs. Example Data
- input.exclude.txt - vSampler only contains all variants that locates on autosomes with MAF > 0.01 based on 1000 genome phase 3 project genotype data, query variants out of this scope will be exclude. Example Data
Detailed options
Program to sampling dataset, the following options are relevant to Sampler
| Option | Description |
|---|---|
| -query-file,-Q:TagArgument |
Path of query file (support plain text and gzip compressed file). Required. TagArgument: arguments with tag and attributes Usage: -I: Possible Tags: {vcf, vcfLike, coordOnly, coordAllele, tab} Possible attributes for all tags: {sep, ci} Possible attributes for "tab" tag: {c, b, e, ref, alt, 0} Attributes should be used with tag, description of attributes: |
| --has-header,-header:Boolean | Indicate whether the first line of input is header line. Default value: false. Possible values: {true, false} |
| --Database,-D:File | The database file. Required. |
| --OutPath,-O:String | The output folder path. Default value: null. |
| --GeneInDis,-GP:GeneInDis | Physical distance cutoff to define gene density of variants. (KB100 means distance in 100KB, KB200 means distance in 200KB, KB300 means distance in 300KB, KB400 means distance in 400KB, KB500 means distance in 500KB, KB600 means distance in 600KB, KB700 means distance in 700KB, KB800 means distance in 800KB, KB900 means distance in 900KB, KB1000 means distance in 1M). Default value: KB500. Possible values: {KB100, KB200, KB300, KB400, KB500, KB600, KB700, KB800, KB900, KB1000} |
| --GeneInLD,-GLD:LD | LD cutoff to define gene density of variants. (LD1 means ld>0.1, LD2 means ld>0.2, LD3 means ld>0.3, LD4 means ld>0.4, LD5 means ld>0.5, LD6 means ld>0.6, LD7 means ld>0.7, LD8 means ld>0.8, LD9 means ld>0.9). Default value: LD5. Possible values: {LD1, LD2, LD3, LD4, LD5, LD6, LD7, LD8, LD9} |
| --inLDvariants,-LDB:LD | LD cutoff to define in LD variants. (LD1 means ld>0.1, LD2 means ld>0.2, LD3 means ld>0.3, LD4 means ld>0.4, LD5 means ld>0.5, LD6 means ld>0.6, LD7 means ld>0.7, LD8 means ld>0.8, LD9 means ld>0.9). Default value: LD5. Possible values: {LD1, LD2, LD3, LD4, LD5, LD6, LD7, LD8, LD9} |
| --MAFDeviation,-MD:MAFDeviation | Deviation range of MAF. Input variant MAF ± MAF deviation range. Required. (D1 means ±0.01, D2 means ±0.02, D3 means ±0.03, D4 means ±0.04, D5 means ±0.05, D6 means ±0.06, D7 means ±0.07, D8 means ±0.08, D9 means ±0.09, D10 means ±0.1) Default value: D5. Possible values: {D1, D2, D3, D4, D5, D6, D7, D8, D9, D10} |
| --disDeviation,-DD:Integer | Deviation range of distance to closest transcription start site (DTCT). Input variant DTCT ± DTCT deviation range. Default value: 5000. |
| --geneDeviation,-GD:Integer | Deviation range of gene density number. Default value: 5. |
| --inLDvariantsDeviation,-LDD:Integer | Deviation range of in LD variants number. Default value: 50. |
| --CellType,-CT:CellType | Roadmap cell type. This should be supplied with `-M,--Mark` Default value: null. Possible values: {E001, E002, E003, E004, E005, E006, E007, E008, E009, E010, E011, E012, E013, E014, E015, E016, E017, E018, E019, E020, E021, E022, E023, E024, E025, E026, E027, E028, E029, E030, E031, E032, E033, E034, E035, E036, E037, E038, E039, E040, E041, E042, E043, E044, E045, E046, E047, E048, E049, E050, E051, E052, E053, E054, E055, E056, E057, E058, E059, E061, E062, E063, E065, E066, E067, E068, E069, E070, E071, E072, E073, E074, E075, E076, E077, E078, E079, E080, E081, E082, E083, E084, E085, E086, E087, E088, E089, E090, E091, E092, E093, E094, E095, E096, E097, E098, E099, E100, E101, E102, E103, E104, E105, E106, E107, E108, E109, E110, E111, E112, E113, E114, E115, E116, E117, E118, E119, E120, E121, E122, E123, E124, E125, E126, E127, E128, E129} |
| --Mark,-M:Marker | Roadmap cell type specific epigenomic mark. This should be supplied with `-CT,--CellType` Default value: null. Possible values: {DNase, H3K4me1, H3K4me3, H3K36me3, H3K27me3, H3K9me3} |
| --Tissue,-TS:TissueType | Match eQTL in tissue. Default value: null. Possible values: {ADIPOSE_SUBCUTANEOUS, ADIPOSE_VISCERAL_OMENTUM, ADRENAL_GLAND, ARTERY_AORTA, ARTERY_CORONARY, ARTERY_TIBIAL, BRAIN_AMYGDALA, BRAIN_ANTERIOR_CINGULATE_CORTEX_BA24, BRAIN_CAUDATE_BASAL_GANGLIA, BRAIN_CEREBELLAR_HEMISPHERE, BRAIN_CEREBELLUM, BRAIN_CORTEX, BRAIN_FRONTAL_CORTEX_BA9, BRAIN_HIPPOCAMPUS, BRAIN_HYPOTHALAMUS, BRAIN_NUCLEUS_ACCUMBENS_BASAL_GANGLIA, BRAIN_PUTAMEN_BASAL_GANGLIA, BRAIN_SPINAL_CORD_CERVICAL_C1, BRAIN_SUBSTANTIA_NIGRA, BREAST_MAMMARY_TISSUE, CELLS_CULTURED_FIBROBLASTS, CELLS_EBV_TRANSFORMED_LYMPHOCYTES, COLON_SIGMOID, COLON_TRANSVERSE, ESOPHAGUS_GASTROESOPHAGEAL_JUNCTION, ESOPHAGUS_MUCOSA, ESOPHAGUS_MUSCULARIS, HEART_ATRIAL_APPENDAGE, HEART_LEFT_VENTRICLE, KIDNEY_CORTEX, LIVER, LUNG, MINOR_SALIVARY_GLAND, MUSCLE_SKELETAL, NERVE_TIBIAL, OVARY, PANCREAS, PITUITARY, PROSTATE, SKIN_NOT_SUN_EXPOSED_SUPRAPUBIC, SKIN_SUN_EXPOSED_LOWER_LEG, SMALL_INTESTINE_TERMINAL_ILEUM, SPLEEN, STOMACH, TESTIS, THYROID, UTERUS, VAGINA, WHOLE_BLOOD} |
| --RegionMatch,-RM:Boolean | Indicator to match variant region or not. The types of variant region are exonic + splicing altering, noncoding and others. Default value: false. Possible values: {true, false} |
| --GCType,-GCT:GCType | Distance range to compute GC content(BP100 means ±100bp, BP200 means ±200bp, BP300 means ±300bp, BP400 means ±400bp, BP500 means ±500bp). Default value: null. Possible values: {BP100, BP200, BP300, BP400, BP500} |
| --GCDeviation,-GCD:GCDeviation | Deviation range of GC. Input GC content ± GC deviation range. (D1 means ±0.01, D2 means ±0.02, D3 means ±0.03, D4 means ±0.04, D5 means ±0.05, D6 means ±0.06, D7 means ±0.07, D8 means ±0.08, D9 means ±0.09, D10 means ±0.1) Default value: D5. Possible values: {D1, D2, D3, D4, D5, D6, D7, D8, D9, D10} |
| --isCrossChr,-CC:Boolean | Indicator of sampling across chromosomes or not. Default value: false. Possible values: {true, false} |
| --excludeInput,-EI:Boolean | Indicator to exclude input SNPs from matched SNPs or not. Default value: true. Possible values: {true, false} |
| --vriantTypeSpecific,-VFS:Boolean | Indicator of doing variant type specific sampling or not. i.e. sample indels for indels, snps for snps Default value: true. Possible values: {true, false} |
| --controlNumber,-SN:Integer | Sample control number Default value: 1. |
| --annoNumber,-AN:Integer | Annotation number Default value: 1. |
| Seed | Random seed. Default value: -1. |
Other vSampler utilities (for advanced usages)
Note
There is no need to
User could download database of
BuildDatabase
The following standard options are relevant to Build Database:
| Option | Description |
|---|---|
| --Config,-C:File | The configuration file that defines the database paths. Required. e.g. eur.db.ini |
| --Population,-P:Population | Population to select. Possible values: {EUR, EAS, AFR, AMR, SAS}. Default value: EUR. Optional. |
| --Thread,-T:Integer | Threads to run the program. Default value: 4. |
Example: this example will build a database named
java -jar -Xmx4g -Xms4g VariantSampler-x.y.z.jar BuildDatabase -C data/eur.db.ini -P EUR -T 4
BuildIndex
To build index for database, The following standard options are relevant to Build Index:
| Option | Description |
|---|---|
| --Database,-D:File | The database file. Required. |
Example:
java -jar -Xmx4g -Xms1g VariantSampler-x.y.z.jar BuildIndex -D EUR.gz
vSampler Databases
Database construction
Genotype call sets of
- URL: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/;
- Release date: 05/02/2013;
- Sample size: EUR: 495, EAS: 497, AFR: 645
Annotation database
First split multi-allelic variants into multiple bi-allelic variants and then left-aligned and normalized reference and alternative alleles of all variants. Duplicate variants that map to the same position with identical reference and alternative alleles were removed.
Sampling database
The number of variants in the annotation database was too large to be feasible for the sampling process. Kept only variants with MAF > 0.01 of the annotation database to construct the sampling database.
Number of variants in vSampler database
Number of variants in vSampler database (
| Super populations | Annotation database | Sampling database |
|---|---|---|
| EUR (European) | 81,647,035 | 9,808,459 |
| AFR (African) | 81,647,035 | 16,750,259 |
| EAS (East Asian) | 81,647,035 | 8,668,864 |
| AMR (Ad Mixed American) | 81,647,035 | 11,184,049 |
| SAS (South Asian) | 81,647,035 | 10,264,032 |
Number of variants in vSampler database (
| Super populations | Annotation database | Sampling database |
|---|---|---|
| EUR (European) | 78,122,255 | 8,873,459 |
| AFR (African) | 78,122,255 | 14,399,202 |
| EAS (East Asian) | 78,122,255 | 7,892,804 |
| AMR (Ad Mixed American) | 78,122,255 | 9,464,290 |
| SAS (South Asian) | 78,122,255 | 9,124,174 |
Note
vSampler kept only variants with
vSampler only allows to match SNPs and Indels that located on autosomes
Annotation of variant properties
MAF : variants’ MAF of EUR, EAS, AFR, AMR and SAS population were computed based on allele frequency information from 1000 Genomes Project phase 3 release as described in database construction section.Distance to closest transcription start site(DTCT) : all 5’ transcription start sites were defined according to GENCODE v32 and then we calculated variants’ distance to the closest 5’ transcription start sites.Gene density : gene density refers to number of genes overlapping with variant loci. Genes were extracted from GENCODE v32, and variant loci were defined by LD thresholds (r2 > {0.1, 0.2, …, 0.9}) or physical distance (window size of 100, 200, ..., 1000 kb).Number of variants in LD : number of variants in LD were calculated using LD thresholds (r2 > {0.1, 0.2, …, 0.9}).GC content : GC content of variants were computed with various window sizes (100bp, 200bp,...,500bp) based on 5 base GC content file hg19.gc5Base.txt.gz from UCSC Genome Browser (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/gc5Base/hg19.gc5Base.txt.gz).Cell type specific epigenomic marks : annotation of cell type-specific epigenomic marks is binary indicator of whether variants fall within selected cell type-specific epigenomic marks. The cell type specific epigenomic marks included DNase I hypersensitive sites (DHSs) and histone modificationsH3K4me1 ,H3K4me3 ,H3K36me3 ,H3K27me3 ,H3K9me3 downloaded from Roadmap Epigenomics Project (https://egg2.wustl.edu/roadmap/web_portal/).eQTL : annotation of eQTL significance is binary indicator of whether variants are significant eQTL variants. Significant eQTL variants data of49 tissues/cell types were downloaded from GTEx project v8 (https://storage.googleapis.com/gtex_analysis_v8/single_tissue_qtl_data/GTEx_Analysis_v8_eQTL.tar). Significance of eQTL variants were determined based on permutation by GTEx project.Coding/noncoding region : We first identified variant effects using Jannovar, and then variants effects were classified as coding or non-coding.
FAQ
Can I use genotype data of sub-populations or populations other from EUR, EAS, AFR, AMR and SAS ?
Yes, but you have to follow aforementioned procedures buildDatabase and buildIndex to index genotype data and annotation databases first. Dependent annotation files can be downloaded from https://1drv.ms/f/s!Aurnn0fjCLv3glA_EoGK_N-daIwo?e=rKm9bh.
Can I run vSampler on my laptop? Is it time consuming to run a complete vSampler process?
Normally, vSampler run well and fast with >=4 GB RAM memory. Hence current laptop are certainty affordable for running vSampler. The whole process takes only <10 minutes with 10000 querys. But runtime will increase about two times with
-CCoption.
Links
- 1000 genome project https://www.internationalgenome.org/home
- GENCODE https://www.gencodegenes.org/
- Roadmap epigenomics project http://www.roadmapepigenomics.org/
- GTEx https://www.gtexportal.org/home/
- UCSC Genome Browser 5 base GC content http://hgdownload.cse.ucsc.edu/goldenPath/hg19/gc5Base
Please cite vSampler as follows:
Huang D#, Wang Z#, Zhou Y#, Liang Q, Sham PC, Yao H*, Li MJ*. vSampler: fast and annotation-based matched variant sampling tool. Bioinformatics. 2021 Jul 27;37(13):1915-1917.